Read about upcoming changes to UnitedHealthcare coverage.
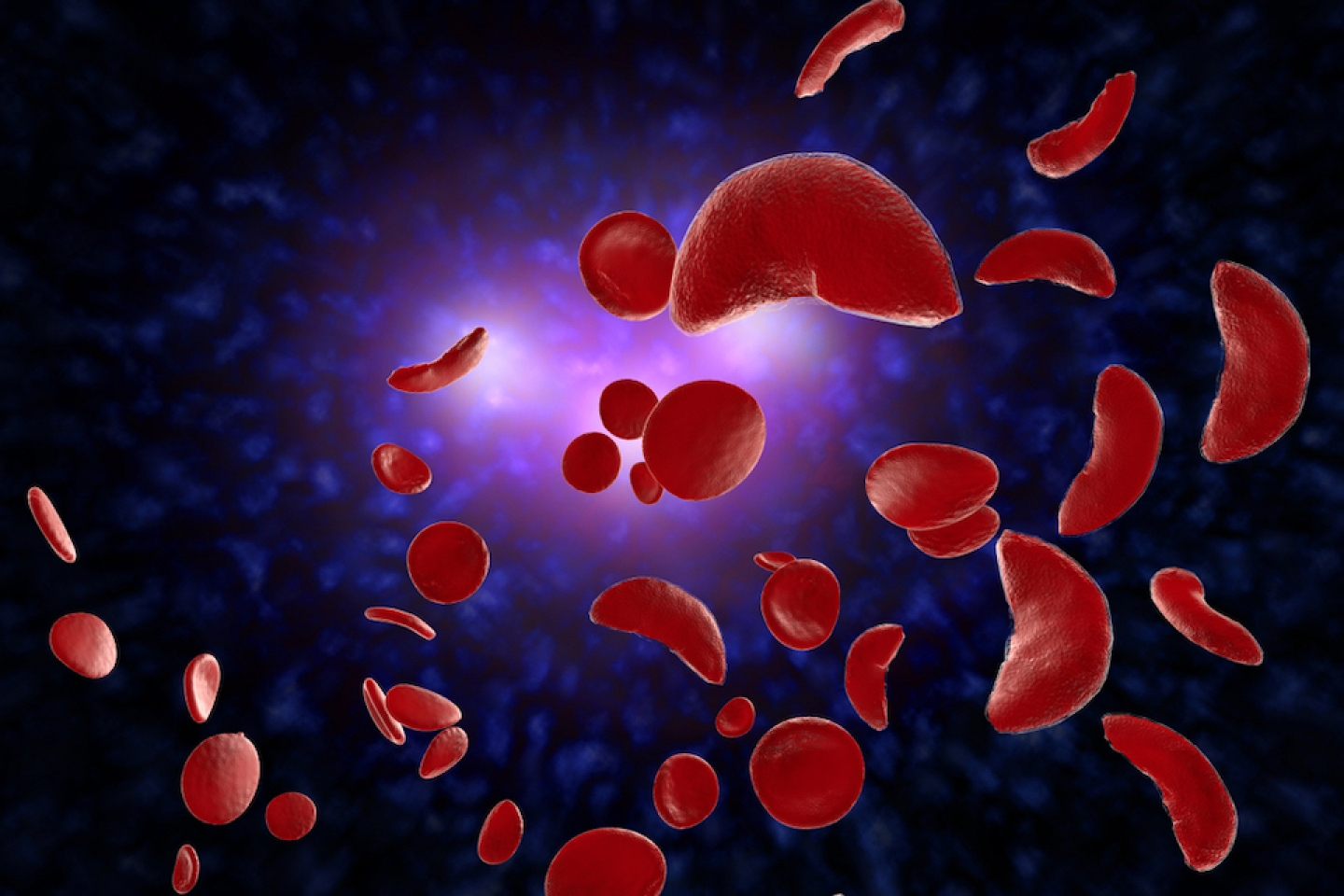
As its name suggests, sickle- or crescent-shaped blood cells characterize sickle cell disease (SCD), a commonly inherited blood disorder that affects the body’s red blood cells.
SCD is an inherited condition, one that a person is born with, says Cheryl Mensah, MD, assistant professor of medicine. Although SCD may affect anyone, it occurs most frequently in people of African, Latin American, or Southeast Asian descent, Dr. Mensah says. “One in 12 African-Americans carries the sickle cell trait, however, one in 16,000 Hispanic-Americans carries the sickle cell trait as well,” she says.
In people with SCD, red blood cells are misshapen and therefore cannot do their job of delivering oxygen to the body. Unlike healthy, oxygen-rich red blood cells, which are round and soft enough to squeeze through tiny blood vessels, those that cause sickle cell disease are C-shaped, hard, and sticky. They neither bend nor move easily. As a result, they block blood flow, depriving the body of oxygen, while also causing pain and other serious problems such as acute chest syndrome, stroke, or eye problems.
Moreover, whereas healthy red blood cells live for about 120 days before new ones replace them, sickle-shaped cells have a far shorter lifespan--only 16 days. This means that patients with SCD have a constant shortage of red blood cells and the oxygen that their bodies need to function properly.
Doctors diagnose SCD with a simple blood test that they usually administer during routine newborn screening. Doctors can also diagnose SCD before birth. Because children with SCD are at an increased risk of infection and other health problems, early diagnosis and treatment are important.
SCD is a lifelong illness, but different types of the disease bring different life expectancies, Dr. Mensah explains. “Patients with sickle cell thalassemia can potentially live to 50-70 years of age, while patients with the most severe form of SCD, known as SS disease, typically have a shorter life expectancy and can live to be 40-50 years old,” she says.
Currently, the only cure for SCD is a bone marrow transplant, a high-risk procedure that entails infusing a patient with a donor’s healthy blood-forming stem cells. For the transplant to work, a donor’s bone marrow must match closely with that of the patient. Siblings are usually ideal donors.
Weill Cornell Medicine’s Center for Blood Disorders offers such transplants, as well as blood transfusions, and treatments with medications, such as crizanlizumab and hydroxyurea, to reduce hospitalization and emergency room visits. “The life expectancy for SCD has increased in the last 20 years and it’s due to the way the specialists at Weill Cornell Medicine manage it, which includes treatment with a medication call hydroxyurea,” Dr. Mensah says.
If you or someone you love has received a diagnosis of SCD, contact WCM’s Center for Blood Disorders. Our expert team of physicians and healthcare practitioners is dedicated to caring for people with all types of blood disorders, including those affecting the red blood cells, white blood cells, platelets, and the clotting system.
Whether you’re newly diagnosed or have previously received care for your blood disorder, we will provide you with the most advanced diagnostic technologies, treatment, and care in a supportive and collaborative environment.